Presentación de caso
Craneofaringioma nasofaríngeo
Nasopharyngeal Craniopharyngioma
Zaida
María León Castellanos1*
https://orcid.org/0000-0002-8463-6588
Glessy Leliebre
Petell1 https://orcid.org/0000-0001-6550-0661
Yicel Torres Harris2
https://orcid.org/0000-0001-6753-3559
Yenicet Rodríguez
Martínez1 https://orcid.org/0000-0002-4111-2645
1Hospital
Pediátrico Sur "Dr. Antonio María Béguez César". Santiago
de Cuba, Cuba.
2Hospital
Militar Docente "Dr. Joaquín Castillo Duany". Santiago de Cuba, Cuba.
*Autor para la correspondencia: zleoncastellanos@gmail.com
RESUMEN:
Introducción:
Los craneofaringiomas son tumores epiteliales raros, localmente agresivos
que habitualmente se localizan en la región selar y supraselar. Derivan
de restos embrionarios del conducto craneofaríngeo. Aunque de naturaleza
benigna, resultan localmente invasivos; infiltran órganos y estructuras
adyacentes como el hipotálamo, la hipófisis, el quiasma y los nervios
ópticos, y originan una considerable morbimortalidad.
Objetivo:
Describir enfermedad infrecuente en niños y modo de actuación.
Caso clínico:
Se presenta caso de niña de 11 años, sin antecedentes patológicos
personales prenatales o perinatales, cuyo síntoma principal fue epistaxis,
hemoptisis ligera y obstrucción nasal derecha, acompañado de cefalea
desde aproximadamente dos meses. Al examen físico no se constatan datos
positivos y se decide llevar al salón de operaciones para explorar nasofaringe
y se observa tumor que ocupa la nasofaringe. Se decide realizar biopsia, donde
el diagnóstico anatomopatológico fue craneofaringioma acantomatoso.
Se le realizan estudios de imagen para valorar la extensión del tumor y
se remite al "Instituto Nacional de Oncología y Radiología" donde
se le realiza exéresis de la lesión por vía transesfenoidal y
tratamiento con radiaciones presentando una evolución satisfactoria.
Conclusion
es: El craneofaringioma es una tumoración infrecuente en edades pediátricas
y el tratamiento curativo incluye combinación de cirugía con radioterapia.
Su pronóstico es favorable si se trata en los primeros estadios.
Palabras clave: tumor supraselar; craneofaringioma; nasofaringe; neoplasia craneal; cirugía.
ABSTRACT
Introduction:
Craniopharyngiomas are rare, locally aggressive epithelial tumors that
are usually located in the sellar and suprasellar regions. They are derived
from embryonic remains of the craniopharyngeal duct. Although benign in nature,
they are locally invasive; they infiltrate adjacent organs and structures such
as the hypothalamus, pituitary, chiasm, and optic nerves, causing considerable
morbidity and mortality.
Objective:
To describe rare disease in children and the approach of action.
Clinical case
report: A case of an 11-year-old girl is reported, she had no personal prenatal
or perinatal pathological history. The main symptoms were epistaxis, slight
hemoptysis and right nasal obstruction, accompanied by headache for approximately
two months. The physical examination did not reveal positive data. It was decided
to take her to the operating room to explore the nasopharynx and a tumor was
observed which occupied the nasopharynx. It was decided to perform a biopsy
that provided a pathological diagnosis of acanthomatous craniopharyngioma. Imaging
studies are performed to assess the extent of the tumor and she was referred
to the National Institute of Oncology and Radiology to remove the tumor by transsphenoidal
surgery and she was treated with radiation. She had a satisfactory evolution.
Conclusions:
Craniopharyngioma is a rare tumor in pediatric ages and curative treatment includes
a combination of surgery and radiotherapy. The prognosis is favorable if it
is treated in the early stages.
Keywords: suprasellar tumor; craniopharyngioma; nasopharynx; cranial neoplasia; surgery.
Recibido:
12/12/2020
Aprobado:14/01/2021
INTRODUCCIÓN
El craneofaringioma es un tumor supraselar o selar, de consistencia quística o sólida. Deriva de restos del ectodermo embrionario. Constituyen entre 1 al 3 % de los tumores intracraneales primarios.(1,2)
Es el tumor más frecuente de la edad pediátrica, histológicamente benigno, pero con carácter expansivo que provoca manifestaciones clínicas secundarias a la afectación de estructuras vecinas,(3,4) tiene comportamiento localmente agresivo por lo que se asocia a menudo a pronóstico desfavorable con frecuentes secuelas neurológicas, visuales y endocrinas.(1,2,5) Son poco frecuentes en niños menores de 2 años de edad, y se diagnostican con mayor frecuencia de 5 a 14 años de edad. No se conocen las causas de estos tumores.(1,2,3,4,5,6) Tienen potencial de malignización bajo, clasificándose como de grado I según la Organización Mundial de la Salud (OMS).(7)
Son tumores poco frecuentes, con incidencia de 0,5-2 casos por millón de personas por año y prevalencia estimada de 1-3 por 100 000 habitantes por año, sin predilección por el sexo. Presentan distribución bimodal, con pico de incidencia entre 5 y 15 años y otro entre la quinta y séptima década de la vida.(1,2,3,7)
Existen dos variedades histológicas: escamoso papilar (15 %) y adamantinomatoso (85 %) que es más frecuente. Este último puede presentarse a cualquier edad, pero predomina en sujetos jóvenes, siendo común en niños (94 %). La variedad papilar es casi exclusiva del adulto.
El diagnóstico del craneofaringioma requiere estudios de imagen, pruebas de laboratorio y estudios histopatológicos. La imagen típica en la tomografía axial computarizada (TAC) en 90 % de los casos, muestra tumor localizado en región supraselar con aspecto quístico, áreas de calcificación y zonas de captación de contraste yodado. Las calcificaciones supraselares son muy sugestivas del craneofaringioma en los niños.(1)
La sintomatología inicial de estos pacientes suele ser muy inespecífica, por lo cual, el periodo de tiempo desde el comienzo de los síntomas hasta la obtención de diagnóstico certero, es de una semana hasta un año.(1,2,7,8). El objetivo de este trabajo es describir una enfermedad infrecuente en niños y su modo de actuación.
CASO CLÍNICO
Paciente femenina, 11 años de edad, procedencia rural, mestiza, sin antecedentes patológicos personales prenatales o perinatales ni familiares. Aparentemente con evaluación nutricional entre 25-50 P (se consideran los valores obtenidos del porcentaje de peso de referencia (peso/peso ideal, P/PI) obtenidos a través de la valoración global objetiva). Es remitida de su área de salud por presentar desde aproximadamente dos meses cuadros a repetición de hemoptisis ligera intermitente y epistaxis, acompañada de cefalea y obstrucción nasal derecha, no presenta fiebre. Se decide su ingreso para estudio y tratamiento. Se le realiza exhaustivo examen físico e interrogatorio a los padres donde no se constatan datos positivos, por lo que es llevada al salón de operaciones para realizar el examen endoscópico, visualizándose tumor que ocupa la nasofaringe del cual se toma biopsia, que informa craneofaringioma acantomatoso.
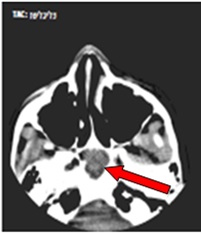
Una vez recibido este diagnóstico, se le realiza TAC para valorar la extensión y características del tumor (Fig.) que informa: imagen isodensa localizada por debajo y detrás del clivus de 38 x 28 mm produciendo desplazamiento de la base del cráneo con aspecto benigno. No presenta alteraciones encefálicas, ni otras alteraciones.
Se remite la paciente al Instituto Nacional de Oncología y Radiobiología (INOR) para mejor estudio y tratamiento, donde se le realiza exéresis de la lesión por vía endoscópica transesfenoidal. Posteriormente recibe tratamiento con radiaciones 30 sesiones de 54 Gy presentando una evolución satisfactoria luego de la terapéutica recibida. Actualmente luego de 6 años de tratamiento, no existen síntomas ni evidencia tumoral, con un seguimiento anual por nuestro centro.
DISCUSIÓN
Los craneofaringiomas son tumores epiteliales raros localmente agresivos que habitualmente se localizan en la región selar y supraselar. Derivan de restos embrionarios del conducto craneofaríngeo formado por la evaginación ectodérmica que constituye la bolsa de Rathke,(1) la cual, en sus inicios, proviene de una evaginación de la orofaringe. Las células del epitelio escamoso remanente de la bolsa de Rathke se pueden extender desde la nasofaringe hasta el tubercinereum dando lugar a que el tumor pueda surgir a partir de remanentes dentro del seno esfenoidal, silla turca o la región supraselar.(1,2) Dichos tumores pueden aparecer en cualquier lugar a lo largo del canal craneofaríngeo, pero lo más frecuente es a nivel supraselar. Otras localizaciones más infrecuentes también han sido detectadas, como en el lóbulo temporal.(1,3)
Puede ser una lesión sólida, quísticas o de naturaleza mixta. Comúnmente calcificados, estrechamente adheridos a las estructuras vasculares y el tejido cerebral adyacente a expensas de nidos celulares infiltrados en la zona de gliosis reactiva peritumoral, 60 % de los tumores tienen ambos componentes.
Los de tipo adamantinomatoso son más prevalentes en niños. Este subtipo de tumor es más quístico y calcificado, más grande en el momento de su presentación y más adherido al tejido neural adyacente como resultado de la fibrosis y la inflamación.(8) Los de tipo escamoso son casi exclusivos de adultos. En más del 70 % de las formas adamantinomatosas se detecta una mutación en el exón 3 del gen de la catenina que no está presente en el craneofaringioma de tipo papilar.(1,2,7,8)
La presentación clínica dependerá de ubicación y tamaño del tumor. Son de crecimiento lento y los síntomas a menudo están presentes en un año o más antes de que el diagnóstico sea establecido, el cual suele ser tardío y tras la aparición de síntomas inespecíficos como dolor de cabeza (por presión intracraneal), déficits visuales, náuseas, vómitos y alteraciones hormonales, estos síntomas, junto con retraso del crecimiento, poliuria y polidipsia son sugestivos de craneofaringioma infantil.(3,8)
Estos pueden causar daños permanentes y severos en las áreas visual, hipotalámica, endocrina y en general en la esfera neurocognoscitiva. El diagnóstico se confirma por procedimientos histopatológicos. Sin embargo, el diagnóstico imagenológico por TAC y Resonancia Magnética (RM), empleando la clasificación imagenológica de Kassam y Puget para este tipo de tumor, son de vital importancia para conocer el grado de respuesta a una eventual cirugía, pues apoyan el planteamiento clínico, tras evaluar las características de la lesión: tamaño, componentes (sólido, quístico o mixto) y extensión hacia estructuras vecinas.(3,8,5,9,10)
Estudios angiográficos (angio-RM, angio-TAC) identifican estructuras vasculares que estén afectadas o desplazadas por el tumor.(2,11)
Los craneofaringiomas que miden más de 2 cm aparecen entre un 14-20 %, los mayores de 4 cm de un 58 a 76 %, entre 2 y 4 cm de un 4-28 % y menores de 2 cm de un 24 a 28 %. La imagen radiológica clásica es una masa sólida (18-39 %) o quístico-sólida (46-64 %) con diversos grados de calcificación (en palomitas de maíz o "en cáscara").(1)
Debido a su ubicación anatómica, los craneofaringiomas son tumores complejos en su tratamiento. Una resección completa tiene riesgos elevados de morbimortalidad, por eso en ocasiones se opta por una resección parcial.(9)
El tratamiento de tumoraciones de esta clase es principalmente quirúrgico, sin embargo, debido a su localización, la resección de estos puede resultar en un procedimiento muy complejo con posibles complicaciones y secuelas. La resección completa es motivo de controversia, pues se ha reportado que en repetidas ocasiones se presentan daños a estructuras contiguas; por otro lado, la resección incompleta ha demostrado la necesidad de reintervenciones y uso de radioterapia postoperatoria. No obstante, en los últimos años. se está optando preferencialmente por la preservación del hipotálamo mediante intervenciones conservadoras, para evitar secuelas provocadas con la cirugía radical.(1,7)
El tratamiento quirúrgico requiere conocimiento detallado de la anatomía de la base del cráneo, así como de diferentes técnicas quirúrgicas. El uso de la endoscopia ha permitido mejor acceso a este tipo de lesiones, disminuyendo las comorbilidades en el paciente y la estadía hospitalaria.(9,12)
El tratamiento quirúrgico puede ser realizado mediante cirugía transcraneal o transesfenoidal, teóricamente, la resección completa debe ser suficiente para curar la enfermedad. La alternativa de tratamiento a la resección es la radioterapia. La radioterapia convencional logra una tasa de supervivencia libre de tumor 95 % a 10 años y 54 % a 20 años, sin embargo, existen técnicas más modernas como la terapia de protones, la radioterapia estereotáctica, radiocirugía y la braquiterapia intracavitaria.(3)
Se concluye que el craneofaringioma es una tumoración infrecuente en edades pediátricas y el tratamiento curativo incluye una combinación de cirugía con radioterapia con un pronóstico favorable si se trata en los primeros estadios.
REFERENCIAS BIBLIOGRÁFICAS
1. Venegas E, Blanco C, Martin T, Soto A. Guía práctica del manejo y tratamiento de los craneofaringiomas y otras lesiones paraselares. Endocrinol Nutr. 2015[acceso 02/10/2020];62(1):e1-e13. Disponible en: https://www.researchgate.net/publication/264640602_Practice_guideline_for_diagnosis_and_treatment_of_craniopharyngioma_and_parasellar_tumors_of_the_pituitary_gland/link/54d490dc0cf25013d0299520/download
2. Valerio J, Sossa A. Craneofaringiomas-Controversias en su manejo quirúrgico. Revisión de la Literatura Mundial. Revista Latinoamericana de Neurocirugía/Neurocirugía. Ecuador. 2019;28(3):41-64.
3. Carracedo D. Craneofaringioma infantil: evaluación y rehabilitación neuropsicológica. [Tesis]. Universidad de Orberta, Cataluny; España. 2020 [acceso 01/10/2020] Disponible en: http://openaccess.uoc.edu/webapps/o2/bitstream/10609/121527/1/dcarracedoTFM0720.pdf
4. Carvajal F. Tratamiento con hormona de crecimiento y craneofaringioma: presentación de caso. revistas.intec.edu. Ciencia y Salud. 2018 [acceso 19/03/2019];2(1). Disponible en: https://repositoriobiblioteca.intec.edu.do/handle/123456789/2703
5. Sánchez I, Pérez R, Ortiz M, Aguilar M, Quevedo N, González P. Correlación de evaluaciones clínicas e imagenológicos en casos con craneofaringioma: Hospital "Hermanos Ameijeiras". 2020 [acceso 01/10/2020]; 5(8):806-18. Disponible en: https://doi.org/10.19230/jonnpr.3418
6. Villarejo F, Martínez J. Tumores cerebrales en niños. Sociedad Española de Pediatría extrahospitalaria y atención primaria (SEPEAP). Rev Pediatr Integral. 2012;XVI(6):475-86.
7. Guanuche K, Flores A, León E, Ceme J, Jimenez W. Craneofaringioma infantil: reporte de un caso. Ecuador. Revista Ocronos. 2019 [acceso 01/02/2020]. Disponible en: https://revistamedica.com/craneofaringioma-infantil/
8. López O, Ortiz M, Cruz P, Salcido Y, Gutiérrez P. Abordaje endoscópico simultáneo endonasal y subtemporal para un craneofaringioma gigante: reporte de un caso. Anales de la Academia de Ciencias de Cuba. 2020 [acceso 02/10/2020];10(1):754. Disponible en: http://revistaccuba.sld.cu/index.php/revacc/article/view/754
9. Ajler P, Castro F, Massa D. Craneofaringiomas: cirugía trans nasal vía endoscópica. Servicio de Neurocirugía, Hospital Italiano de Buenos Aires, Argentina. Rev Argent neuroc. 2020 [acceso 02/10/2020];34(1):42-44. Disponible en: https://www.ranc.com.ar/index.php/revista/article/view/27/48
10. Müller H. Craniopharyngioma. Nat. Rev. Dis. Primers. 2019 [acceso 02/10/2020];75(5). Disponible en: https://www.researchgate.net/deref/http%3A%2F%2Fdx.doi.org%2F10.1038%2Fs41572-019-0125-9
11. Dinza E, Martínez J, Pons L, García O. Resonancia magnética en pacientes con tumores más frecuentes en la región selar. Medisan. 2017 [acceso 02/10/2020];21(6):725-730. Disponible en: http://scielo.sld.cu/pdf/san/v21n6/san13216.pdf
12. López O, Ortiz M, Cruz P, Salcido Y, Gutiérrez P, Ortiz M. Abordaje endoscópico simultáneo endonasal y subtemporal para un craneofaringioma gigante: reporte de un caso. Anales de la Academia de Ciencias de Cuba. 2020 [acceso 02/10/2020];10(1):754. Disponible en: http://revistaccuba.sld.cu/index.php/revacc/article/view/754
CONFLICTO DE INTERESES
Los autores declaran que no existe conflicto de intereses de ningún tipo.
CONTRIBUCIONES DE LOS AUTORES
Zaida María León Castellanos: Presentó la idea original del trabajo, realizó el estudio y tratamiento del caso, revisó la literatura.
Glassy Leliebre Petell: Realizó el estudio y tratamiento del caso, revisó la literatura.
Yicel Torres Harris: Realizó el estudio y tratamiento del caso, revisó la literatura.
Yenicet Rodríguez Martínez: Revisó la literatura, realizó informe final del texto.