Presentación de casos
Displasia cleidocraneal
Cleidocranial dysplasia
Darselys
Rivero Linares1*
https://orcid.org/0000-0002-8674-1031
Orlando Capote Tabares2
https://orcid.org/0000-0003-1999-6373
1Hospital
General Docente "Enrique Cabrera." La Habana, Cuba.
2Hospital
Pediátrico Universitario "Borrás-Marfán." La Habana, Cuba.
*Autor para la correspondencia: darselysrl@gmail.com
RESUMEN
Introducción:
La disostosis cleidocraneal es un síndrome craneofacial de baja incidencia
cuyas características son estatura baja, anomalías craneofaciales,
claviculares, vertebrales y de la región pélvica. Presenta una etiología
de transmisión autosómica dominante, produce efecto de alargamiento
del cuello y estrechez de los hombros, nariz de base ancha y puente nasal hundido,
quistes y múltiples dientes supernumerarios; el coeficiente de inteligencia
es normal.
Objetivo:
Presentar dos casos interesantes de displasia cleidocraneal.
Casos clínicos:
El primer caso es un paciente de 28 años de edad, sexo masculino, que no
se le han caído los dientes temporales. Al examen regional se observa presencia
de hipertelorismo, cráneo en forma de corazón, puente nasal deprimido
y nariz ancha, entre otras alteraciones. El segundo caso es la madre del paciente
con características clínicas semejantes a las ya descritas.
Conclusiones:
La disostosis cleidocraneal es un síndrome poco frecuente, que tiene un
patrón de herencia autosómico dominante. El diagnóstico debe
hacerse oportunamente para tomar acciones médico- quirúrgicas que
mejoren la calidad de vida de estos pacientes, su manejo debe ser multidisciplinario,
ya que al presentar anomalías en varios sistemas deben ser evaluados por
ortopédicos, otorrinolaringólogos, genetistas, como parte de un manejo
integral de estos casos.
Palabras clave: displasia cleidocraneal; alteraciones óseas; retenciones dentarias múltiples.
ABSTRACT
Introduction:
Cleidocranial dysostosis is a low incidence craniofacial syndrome
whose characteristics are short stature, craniofacial, clavicular, vertebral
and pelvic region anomalies. It shows autosomal dominant transmission etiology,
it produces lengthening of the neck and narrowing of the shoulders, broad-based
nose and sunken nasal bridge, cysts and multiple supernumerary teeth; IQ is
normal.
Objective:
To present two interesting cases of cleidocranial dysplasia.
Clinical
cases: The first case we report here is a 28-year-old male patient,
whose primary teeth have not fallen out. The regional examination showed the
presence of hypertelorism, heart-shaped skull, depressed nasal bridge and wide
nose, among other alterations. The second case is this patient's mother with
similar clinical characteristics already described.
Conclusions:
Cleidocranial dysostosis is a rare syndrome that has an autosomal dominant inheritance
pattern. The diagnosis must be made in a timely manner to take medical-surgical
actions that improve the quality of life of these patients, their management
must be multidisciplinary, since there are anomalies in several systems that
must be assessed by orthopedia, ENTa, genetics, as part of a management comprehensive
of these cases.
Keywords: cleidocranial dysplasia; bone alterations; multiple dental retentions.
Recibido:
12/09/2020
Aceptado: 17/11/2020
INTRODUCCIÓN
Las displasias esqueléticas, también conocidas como osteocondrodisplasias, son un grupo de trastornos del desarrollo que se caracterizan por anormalidades en el crecimiento y el mantenimiento del hueso y el cartílago.(1,2) Entre estas alteraciones se puede encontrar la Displasia Cleidocraneal (DCC) que es una afección descrita por primera vez en 1766 por Morand y posteriormente en 1897 por Pierre Marie y Sainton que la nombran "disostosis cleidocraneal". Es también conocida en la literatura médica con los nombres: Enfermedad de Marie Sainton,Sindrome de Scheuthauer Marie Sainton y disostosis mutacional.(2)
Etiología
Aunque se describe herencia autosómica dominante en la mayor parte de los casos, unos pocos siguen una herencia recesiva. La forma dominante responde a mutaciones del gen CBFA1/RUNX2 (6p21) del factor de transcripción CBFA1, encargado de la diferenciación osteoblástica. Lo que supone una alteración preferente de la osificación membranosa que produce, ante todo, defectos craneales, claviculares y pélvicos. Otros genes mutados implicados en esta patología son el MSX2 (5q34-35) que cursa con hipoplasia clavicular. (3,4)
Este gen controla la diferenciación osteoblástica de las células precursoras y es esencial en la formación de hueso endocondral y membranoso. También se ha vinculado con la morfogénesis dentaria.(5)
Epidemiología
Es considerada como una displasia esquelética, corresponde a una enfermedad de origen genético, muy poco frecuente en la población, con una prevalencia de 1/1.000.000 habitantes. (4)
Características generales
Los defectos óseos en pacientes con DCC implican principalmente las clavículas, el cráneo y la mandíbula, si bien se pueden encontrar en otros huesos una amplia variedad de anomalías. Las clavículas están ausentes, uni o bilateralmente, en alrededor del 10 % de todos los casos. Más comúnmente, las clavículas muestran diversos grados de hipoplasia y malformaciones. En algunos casos, el paciente puede aproximar los hombros delante del pecho, esto es lo más característico y patognomónico del síndrome. El paciente muestra cuello largo y generalmente es de baja estatura. (5,6)
Características de cráneo y cara
En la cabeza se destaca un cráneo grande y braquicéfalo, el abombamiento frontoparietal con suturas y fontanela anterior amplias; son frecuentes los huesos wormianos. Los senos paranasales y las celdillas mastoideas tienen un desarrollo tardío o incompleto; los huesos esfenoidales son pequeños.
En la cara se denota hipertelorismo y ligero exoftalmos. Las anomalías dentarias presentes son: dientes supernumerarios, mala implantación, mala oclusión y posibles alteraciones estructurales (hipoplasia del esmalte, caries precoz, quistes de retención, aplasias); el paladar es ojival (a veces con fisura palatina), hay presencia de hipertrofia gingival.(6,7)
CASOS CLÍNICOS
Se presenta a consulta caso clínico de paciente de 28 años de edad, sexo masculino, con la preocupación de que no se le han caído los dientes temporales. Al interrogatorio se conoce que la madre presenta características fenotípicas similares con el antecedente de diagnóstico de Displasia Cleidocraneal e igual historia de retenciones dentales.
Caso 1 Hijo
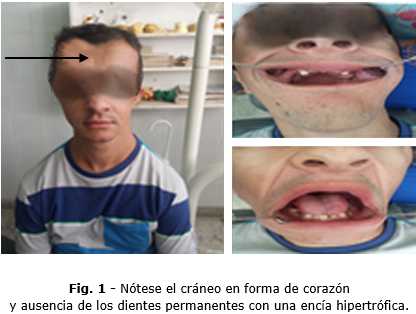
Al examen físico general se observan otras anomalías como baja estatura, movilidad anormal de los hombros, hombros caídos, tórax acampanado, hipertricosis. En el examen regional observamos presencia de hipertelorismo, cráneo en forma de corazón, puente nasal deprimido, nariz ancha, tercio medio e inferior más pequeños con relación al tercio superior. Y al examen intraoral se constata la persistencia de algunos dientes temporales y ausencia de los dientes permanentes con una encía hipertrófica que impide palpar la posible presencia de dientes retenidos (Fig. 1).
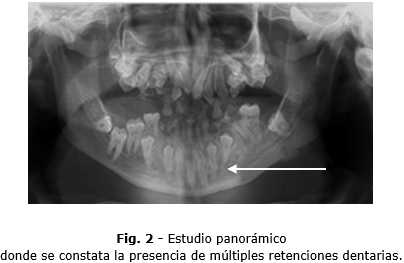
Se realiza estudio panorámico a este paciente donde constatamos la presencia de múltiples retenciones dentarias (Fig. 2).
Se procedió al tratamiento quirúrgico con la exéresis de todos los dientes en 4 cirugías con anestesia local y 2 con anestesia general donde se extrajeron un total de 57 dientes entre temporales y permanentes. El paciente fue rehabilitado con una prótesis mucosoportada mejorando su estética y función masticatoria, así como el habla.
Caso 2 Madre
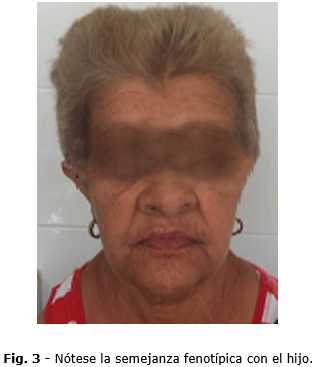
La paciente presenta semejantes características típicas del síndrome como baja estatura, movilidad anormal de los hombros, hipertelorismo. El cráneo acorazonado (Fig. 3). En su examen intraoral presenta retención dentaria de los terceros molares inferiores y persistencia de dientes con anatomía que no se corresponde a su posición.
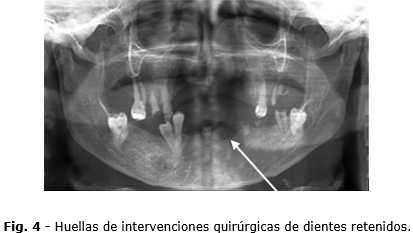
En la radiografía panorámica (Fig. 4) realizada se observa en el hueso las huellas de dientes retenidos extraídos en múltiples cirugías anteriores.
Se le realizó la exéresis de los dientes restantes y se rehabilitó con una prótesis mucosoportada mejorando su función masticatoria y la estética.
DISCUSIÓN
La esqueletogénesis es el proceso de origen, formación y desarrollo del esqueleto que requiere una estrecha interacción entre diversos mecanismos reguladores que controlan la determinación y diferenciación celular, la orquestación de genes específicos de hueso y de cartílago y otros modificadores y la influencia de las interacciones célula-célula y célula-matriz. Desde el punto de vista embriológico, cada osteocondrodisplasia es el resultado de una alteración en cualquiera de estos mecanismos, sea por un producto celular anormal o por un desbalance del desarrollo, generado por anomalías cromosómicas.(8)
Sin embargo, no siempre hay una clara correlación entre el defecto genético y los fenotipos clínicos en todos los casos, lo que indica que pueden estar involucrados otros factores, incluso epigenéticos, reguladores y ambientales, con una prevalencia que oscila entre 1,1 y 7,6 por cada 10 000 nacimientos. No hay diferencias en la prevalencia de la enfermedad según etnia o género.(2,8)
Las características faciales incluyen: frente prominente, hipertelorismo, hipoplasia mediofacial, puente nasal deprimido y paladar alto. Radiológicamente, se puede encontrar un retraso en el cierre de las suturas sagital y metópica y de las fontanelas; así mismo, senos paranasales subdesarrollados entre otras.(5,9)
También es importante identificar la pérdida de audición, que puede deberse a cambios estructurales y funcionales de los huesos temporales y propios del oído, junto con la formación inusual o anormal del paladar o disfunción de la trompa de Eustaquio, donde aparecerían trastornos conductivos, que puede requerir la colocación de tubos de ventilación, sin embargo, también la hipoacusia puede deberse a la función anormal de los nervios auditivos y encontrar presencia de curvas audiométricas neurosensoriales. Por lo tanto, es preciso que un otorrinolaringólogo evalúe a los pacientes de manera rutinaria y que sean sometidos a pruebas audiológicas.(7,10)
El manejo debe ser individualizado, según las necesidades de cada paciente, derivadas de su fenotipo, son fundamentales las del punto de vista estético y funcional. Las anomalías dentales son un aspecto importante en el tratamiento de estos pacientes, algunos requerirán múltiples tratamientos para extracción de dientes supernumerarios y lograr un correcto alineamiento dental. La cirugía craneofacial puede ser necesaria para corregir las anomalías del cráneo.
La consejería genética de la displasia se basa en la herencia autosómica dominante, en la cual el riesgo para los pacientes de tener hijos afectados es del 50 %. Cabe aclarar que esta es una enfermedad que varía en su presentación clínica y que sus principales complicaciones son de orden dental y otorrinolaringológicas, las cuales deben ser intervenidas de modo temprano. Además, pese a ser una enfermedad de carácter sistémico, tiene un buen pronóstico, y solo en casos esporádicos relacionados con re-arreglos cromosómicos se asocia a déficit cognitivo.
En ninguno de los dos pacientes presentados la enfermedad estuvo asociada a un déficit cognitivo, ni a retraso del desarrollo psicomotor.
Los estudios moleculares permiten conocer los genes implicados en la diferenciación osteoblástica y el desarrollo de osificación ósea, lo que permite también el diagnóstico, en aquellos casos con variantes clínicas en los cuales los síntomas y la radiología, no permita delinear bien por el fenotipo.(2,9)
Se concluye que, la Disostosis Cleidocraneal es un síndrome poco frecuente, que tiene un patrón de herencia autosómico dominante. El diagnóstico debe hacerse oportunamente para tomar acciones médico-quirúrgicas que mejoren la calidad de vida de estos pacientes, su manejo debe ser multidisciplinario, ya que al presentar anomalías en varios sistemas deben ser evaluados por ortopédicos, otorrinolaringólogos, genetistas, como parte de un manejo integral de estos casos.
REFERENCIAS BIBLIOGRÁFICAS
1. Ortega R, Suárez F. Displasia cleidocraneal: presentación de un caso. Univ Med. Colombia. [acceso 12/07/2020];2016;57(1):115-22. Disponible en: http://10.11144/Javeriana.umed57-1.dcpc .
2. Delys R, Escudero R, Pérez A. Síndrome de Marie Sainton. Presentación de casos. Rev Acta Medica del Centro. Cuba. [acceso 05/11/2020];2011:5(2): Disponible en: http://www.actamedica.sld.cu/r2_11/marie_sainton.htm .
3. Briggs M, Bell P, Pirog K. The utility of mouse models to provide information regarding the pathomolecular mechanisms in human genetic skeletal diseases: The emerging role of endoplasmic reticulum stress (Review). Int J Mol Med. 2015;35(6):1483-92.
4. Castro A, Escobar E, García G. Displasia cleidocraneal: Revisión y estudio de las características clínicas y radiográficas de una familia chilena. Rev Odontop Latinoam. 2011:1(1).
5. Toro B, Pérez A, Fierro C. Disostosis cleidocraneal: Revisión de la literatura a propósito de un caso clínico. J Oral Res. [acceso 05/11/2020];2012;1(1):22-26.
6. Márquez N, Santana E, Marrero J, Fernández G, Tamayo V. Caracterización de la disostosis cleidocraneal en una familia. Rev Correo Científico Médic, Holguín, Cuba. 2013;17(4).
7. Sánchez C. Disostosis cleidocraneal. Estudio clínico, radiográfico y genético de una familia. Rev Cubana Med. 1999;38(2).
8. Arocha R, Vázquez C, Vázquez A, Cruz R. Disostosis cleidocraneal. Estudio familiar. Rev Cubana Med. 2002 [acceso 11/05/2020];41(3):178-84. Disponible en: http://scielo.sld.cu/scielo.php?pid=S0034-75232002000300010&script=sci_arttext
9. Tanaka J, Ono E, Filho E, Castilho J, Moraes L, Moraes M. Cleidocranial dysplasia: importance of radiographic images in diagnosis of the condition. J Oral Sci. 2006;48(3):161-166.
10. Márquez N, Santana E, Marrero J,Fernández G, Tamayo V. Caracterización de la disostosis cleidocraneal en una familia Correo Científico Médico. Holguín 2013;17(4).
11. Medina O, Muñoz N, Moneriz C. Displasia cleidocraneal: reporte de un caso. Rev. Chil. Pediatría. 2017;88(4):517-23.
Conflicto de intereses
Los autores declaran que no existe conflicto de intereses de ningún tipo.
Contribuciones de los autores
Darselys Rivero Linares: Idea original del trabajo, estudio y tratamiento de los casos, revisión de la bibliografía.
Orlando Capote Tabares: Estudio y tratamiento de los casos, redacción del texto final.